Es gibt ein Jubiläum. Vor ziemlich genau 5 Jahren wurde die MDR (Medical Device Regulation) veröffentlicht. Ja, es sind tatsächlich schon 5 Jahre. Aufgrund von Übergangsfristen, Verschiebungen und Corona mag man das kaum glauben. In dieser Zeit wurde viel diskutiert, dokumentiert und delegiert. In all den Diskussionen über deren Umsetzung kann man schnell mal das Gefühl bekommen, dass es sich bei der MDR um eine MDD (Medical Device Directive) mit extra Inhalt, extra Kapitel und extra Details handelt. Wieso also nicht einfach eine neue Version der MDD, eine MDD 2.0?
Eine Antwort liefert bereits die Namensgebung. „R“ für Regulation und „D“ für Directive. Die MDR ist also eine Verordnung, wohingegen die MDD lediglich eine Richtlinie ist. Der Unterschied liegt im Geltungsbereich.
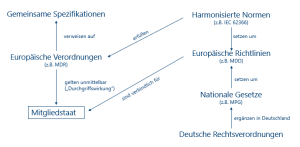
Bis vor 5 Jahren war es so, dass die MDD allein keine rechtliche Verpflichtung zu deren Einhalten beinhaltete. Sie ist eben eine Richtlinie, die zunächst erstmal nur eine gewisse Orientierung und Empfehlung darstellt. Erst durch die nationale „Implementierung“ (in Deutschland durch das Medizinproduktegesetz) erlangt die MDD ihre Gesetzesrelevanz. Jeder europäische Mitgliedstaat musste also der MDD durch seine nationalen Gesetze Gültigkeit verschaffen. Dieser Zwischenschritt ist mit der MDR nicht notwendig, sie gilt unmittelbar. Man spricht hier von einer sogenannten „Durchgriffswirkung“.
Neben der MDD gibt es noch weitere Richtlinien, welche in der regulatorischen Landschaft von Medizinprodukten eine Rolle spielen: Die AIMDD (für aktiv implantierbare medizinische Geräte) und die IVDD (für In-vitro-Diagnostika). Die MDR deckt inhaltlich auch die AIMDD ab. Die IVDD wurde jedoch nicht in die Verordnung integriert. Sie wurde durch eine separate Verordnung ersetzt, der IVDR.

Aber warum nun plötzlich diese Änderung? Auslöser dieser Umstellung war der sogenannte PIP-Skandal. Ein französischer Hersteller für Brustimplantate, Poly Implant Prothèse (PIP), hat in der Produktion Industriesilikon anstelle von medizinischem Silikon verwendet. Diese Implantate reißen schneller und stellen ein hohes Risiko für die betroffenen Frauen dar. Um Patienten und Anwender künftig vor solch kriminellen Machenschaften zu schützen, wurde an zwei Stellschrauben gedreht:
- Direktere Rechtsform
- Verschärfter Inhalt
Neue Rechtsform
Die angepasste Rechtsform erlaubt dem Gesetzgeber nun einen unmittelbaren Einfluss auf die Zulassungsvoraussetzungen und den gesamten Produktlebenszyklus eines Medizinprodukts. Um ein Medizinprodukt auf den Markt zu bringen, muss es sogenannte „grundlegende Anforderungen“ erfüllen. Hersteller müssen deren Einhaltung aktiv nachweisen, ein Bekenntnis allein reicht hier nicht. Und hier kommen nun auch die oftmals geradezu verhassten Normen ins Spiel.
Da die grundlegenden Anforderungen eher unkonkret als stichhaltig formuliert sind, gestaltet es sich für Hersteller nicht gerade einfach, ihrer Beweispflicht nachzukommen. Genau da sollen und können Normen helfen. Sie überführen die unbestimmten Anforderungen in konkrete Bestimmungen. Sie stellen also eigentlich mehr ein Hilfsmittel als ein Hindernis dar. Ja, Normen können kleinlich sein, aber sie definieren eindeutige Kriterien, mit denen man als Hersteller auf der sicheren Seite ist. Zudem steht es theoretisch jedem Hersteller frei, eine eigene Strategie für die Beweispflicht zu entwickeln und umzusetzen.
Dennoch kommt es vor, dass die Normenlandschaft Lücken aufweist und an gewissen Stellen nicht ausreicht. Um diesem Problem Abhilfe zu schaffen, gibt es für die EU-Kommission nun das Instrument der „gemeinsamen Spezifikationen“. Diese sollen die existierenden Normen wo nötig ergänzen. Neben der generellen Durchgriffswirkung der MDR, hat sich die EU-Kommission damit eine zusätzliche Möglichkeit eines direkten Einflusses auf die Zulassung von Medizinprodukten geschaffen.
Neuer Inhalt
Im Zuge der geänderten Rechtsform wurde auch der Inhalt der MDR angepasst, der tatsächlich um einiges mehr umfasst als bisher. Die größten Änderungen finden sich dabei in den Anforderungen an die technische Dokumentation, an die Produkte selbst und in der Klassifizierung von Medizinprodukten.
Eine der größten Änderungen bzgl. Dokumentation stellt die Einführung einer UDI-DI (Unique Device Identification – Device Identification) dar. Dieses sich etwa seltsam wiederholende Akronym verleiht jeder Instanz eines Medizinproduktes des gleichen Typs eine eindeutige Identifikationsnummer, woraus eine schnellere und einfachere Nachverfolgung resultiert.
Eine weitere Verschärfung findet sich in der Post-Market-Surveillance, also dem Überwachen nach dem Inverkehrbringen. Die MDR macht dazu viel konkretere und detailliertere Angaben als die MDD. Gefordert sind unter anderem ein definierter Post-Market-Surveillance-Prozess, eine Systematik zum Konsolidieren, Auswerten und Bewerten von Daten und daraus abgeleitete Maßnahmen wie etwa ein Rückruf oder eine CAPA (corrective and preventive actions).
Die vorangegangenen Beispiele mögen den ein oder anderen Hersteller bereits ins Schwitzen bringen, dennoch müssen wir jetzt noch einmal ganz tapfer sein. Es gibt nämlich eine Änderung in der MDR, die zu den folgenschwersten gehört: Die Klassifizierungsregeln für Software. Diese sind nun so konzipiert, dass es künftig kaum noch Software gibt, die unter die Klasse I fällt. Folglich muss ab sofort für das Inverkehrbringen von medizinischer Software sowohl die benannte Stelle miteinbezogen werden als auch ein Qualitätsmanagementsystem etabliert werden. Gerade für kleinere Hersteller ist diese Änderung mit erheblichen Kosten und auch einem erheblichen zeitlichen Aufwand verbunden.
Fazit
Zusammenfassend kann gesagt werden, dass für Hersteller von Medizinprodukten der hauptsächliche Unterschied von MDR und MDD tatsächlich in dem erweiterten und detaillierteren Inhalt liegt. Ja, dieser ist herausfordernd und sehr zeitintensiv in der Umsetzung. Dabei ist es nur verständlich, dass schnell der Eindruck einer MDD 2.0 erweckt wird. Bei der MDR handelt es sich jedoch nicht um eine MDD + Schnickschnack, da dessen Einordnung in der regulatorischen Landschaft eine signifikant andere ist.
Mehr Informationen zum Thema Medical Software Processes findest du hier in unserem Kompetenzfeld.
- MDD + Schnickschnack = MDR? - 6. Oktober 2022